11 Steps to Performing a Robust Product Risk Analysis
By: Theresa Ramirez, Senior Quality Engineer, San Diego office
In this whitepaper we will provide an overview of the risk management process for a medical device and the important role it plays in bringing safe and effective products to market. We will describe key elements of the process, clarify terminology, and provide best practices for performing a risk analysis of a medical device based upon the internationally recognized standard ISO 14971: Application of Risk Management to Medical Devices. The risk management process described in this document can also be used as guidance for managing risk associated with other product types that are not medical devices, but present safety concerns.
In addition, we will provide editable templates and sample documents that you can use to perform a risk analysis for your next product, along with some sample documents containing actual examples to further your understanding. The sample documents have been created for an electromechanical Infusion Pump medical device and are intended for the purposes of example only. See APPENDIX A Templates and Sample Documents.
OVERVIEW OF RISK MANAGEMENT
Risk associated with a medical device is defined as the combination of the Probability (or likelihood) of occurrence of harm when exposed to a hazard (potential source of harm) and the Severity of that harm; Risk (R)= Severity (S) x Probability (P). See also APPENDIX C for list of key terms and definitions used in the body of this paper.
The use of a medical device involves an inherent degree of risk, even after risks have been reduced to an acceptable level, due to the fact they are used to treat or diagnose patients. Risks associated with a medical device can be related to injury to the patient, the user, and/or other persons. Risks can also be related to damage to property or the environment.
Risk management is the systematic application of management policies, procedures, and practices to the task of identifying, analyzing, evaluating, controlling, and monitoring risk. The risk management process provides the framework within which experience, insight, and judgement can be applied systematically to manage product risk throughout the life cycle of the medical device.
Risk management is not only essential to ensuring a safe and effective medical device but is also a regulatory requirement in many countries, including the US, Canada, and Europe, in order to bring new medical devices to market. The FDA’s Quality Management System (QSR) and the widely recognized international standard ISO 13485 Medical Devices – Quality Management System both require risk management as part of the realization process for medical devices. In addition, the international standards IEC 60601-1 Medical Electrical Equipment – Part 1: General requirements for basic safety and essential performance, and IEC 62304 Medical device software – Software life cycle processes, both require a risk management process compliant with ISO 14971.
ISO 14971 has defined a risk management process for medical devices consisting of the following elements: Risk Analysis, Risk Evaluation, Risk Control, Evaluation of Overall Risk, Risk Management Review and Production and Post-Production Activities. See Figure 1 below for a schematic representation of all these elements. Risk Analysis, Risk Estimation and Risk Control are the core elements of the product risk management process of a medical device and will be the focus of this whitepaper.
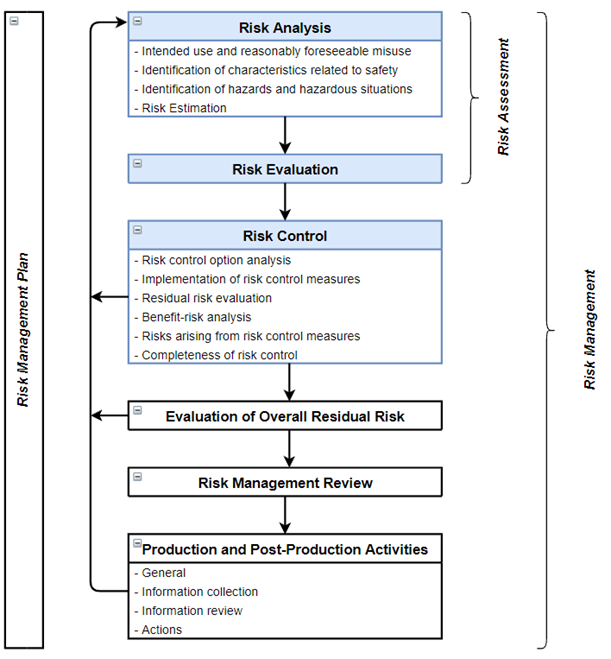
Figure 1: ISO 14971:2019 Risk Management Process
PERFORMING A PRODUCT RISK ANALYSIS
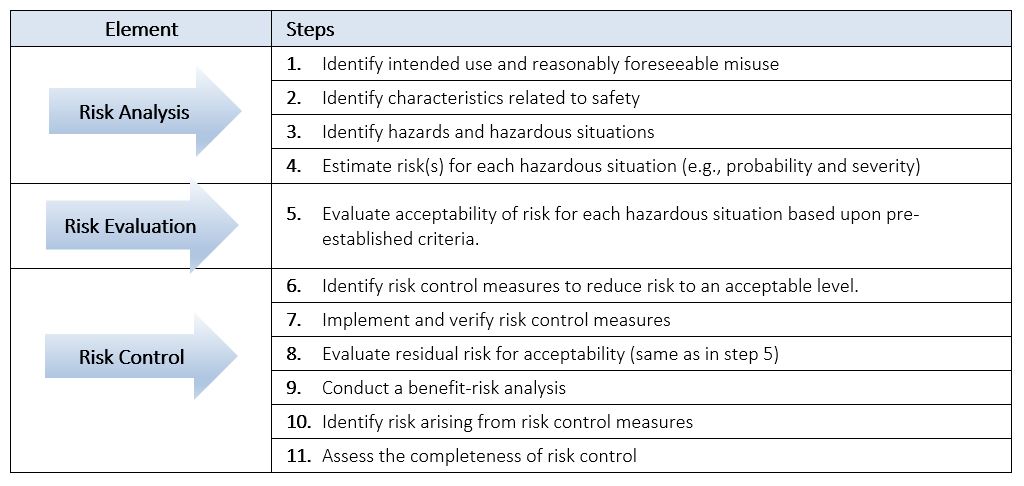
When we talk about “performing” a Product Risk Analysis we are actually referring to performing and documenting not only the Risk Analysis, but also the Risk Evaluation and Risk Control of a device. These three core elements can be summarized into the 11 steps listed in Table 1 below. Examples will be provided throughout this section to guide and help you in performing and documenting a Product Risk Analysis.
Table 1: Steps to Performing a Product Risk Analysis
A risk analysis of a medical device is often referred to as a “hazard” analysis given that the focus of the risk analysis is on safety of the device, and the analysis begins with identifying the hazards associated with the use of the device.
Traceability of risk controls is important in order to demonstrate that all identified hazards and hazardous situations have been addressed in the product prior to shipment. ISO 14971 requires traceability for each identified hazard to the risk analysis (e.g., severity and occurrence), and the evaluation (e.g., estimation of risk), implementation and verification of risk control measure, and the evaluation of residual risk. We have included a Product Risk Analysis Worksheet Template for your use to document your product risk analysis and provide the traceability of each hazard as required by ISO 14971.
Performing a risk analysis requires a cross-functional team that includes experts in the disciplines applicable to the product being developed, including usability and clinical/application specialists that understand the use of the device and potential harms associated with its use.
There are various techniques/tools that can support a risk analysis, among them include a Preliminary Hazard Analysis, Failure Modes Effects Analysis (FMEA), and Fault Tree Analysis (FTA). A full list of risk analysis techniques is described in ISO/TR 14971:2020 Annex B and may be used as needed but will not be covered in this paper.

1. Identify Intended Use and Reasonably Foreseeable Misuse
Before one can begin identifying hazards associated with a medical device, it is critical to first identify and document the intended use and reasonably foreseeable misuse of the device. The intended use of a medical device is often contained in a Use Specification or similar type document and should include:
- The intended medical indication, e.g., treatment or diagnosis of a particular disease or condition
- Patient population, e.g., age groups, gender, or disease state
- Part of the body or type of tissue interacted with, e.g., leg or arm, etc.
- User profile, e.g., patient, lay person, health care provider, service engineer, etc.
- User environment, e.g., home, hospital, intensive care, doctor’s office, ambulance, etc.
- Operating principle (how the treatment or diagnosis is achieved)
Reasonably foreseeable misuse is defined as the use of the medical device in a way that is not intended, but which can result from readily predictable human behavior. Examples of this include use errors (e.g., slip, lapse or mistake), intentional acts of misuse (e.g., non-compliance with operating or user instructions), and intentional use of medical device for other applications than specified or intended (also referred to as off-label use).
2. Identify Characteristics Related to Safety
Another important step in identifying the hazards associated with a medical device is to identify and consider characteristics of the medical device that are related to safety. This can be done by asking a series of questions regarding the intended use, reasonably foreseeable misuse (e.g., intentional or unintentional use of a product or system in a way not intended by the manufacturer), manufacture, and disposal of the device. ISO/TR 24971:2020 Medical devices – Guidance on the application of ISO 14971, provides a list of questions and factors to consider assisting in identifying all characteristics of the medical device that could affect safety. This list is not exhaustive or representative of all medical devices, and some questions may not be applicable to a particular medical device. This list is only meant as a guide and should be applied appropriately and customized as needed for your particular device.
3. Identify Hazards and Hazardous Situations
It is helpful to create a preliminary list of potential hazards and hazardous situations early in the product life cycle in order to quickly identify high risk areas that can be effectively eliminated or reduced by changing the design architecture. Changing the design or moving to an alternative design is much easier to do in the beginning of the project when you have the most potential flexibility to do so.
We have included a Preliminary Hazards List Template to help you in the process of identifying and documenting preliminary hazards and hazardous situations. This template also includes questions on characteristics related to safety from ISO/TR 24971:2020 Annex A. The output of this Preliminary Hazard List will not only serve to inform the early design but will provide input to kick-start the Product Risk Analysis.
Hazard
ISO 14971 defines a hazard as a source of harm and can be present in both normal operation and fault conditions. For example, high voltage, falling objects, static discharge, toxins, are all potential sources of harm depending upon the type of device and its use.
A failure mode is often mistakenly confused as a hazard, but a failure mode is a source of the harm. However, a failure mode has the potential to result in a hazardous situation that leads to harm.
Identify all potential hazards known and foreseeable, associated with your product. Do this using the information gathered from steps 1 and 2 above, along with other available sources of information. Sources of information may include relevant clinical research, applicable regulations and safety standards, post-production data, complaint history and publicly available information about similar products.
ISO 14971 also provides guidance on the different types of hazards that may be encountered, such as: energy hazards (e.g., electrical, mechanical, acoustic, thermal, etc.), biological hazards (e.g., bacteria, viruses, toxins, etc.) and performance hazards (e.g., functionality, information, data, etc.). This is a good place to start but is not necessarily a complete list, nor will all of the hazards be applicable to your device. It is important to identify only those types of hazards that are applicable to the device being analyzed.
Hazardous Situation
A hazardous situation occurs when people, property, or the environment are exposed to one or more hazards. A hazard by itself does not cause harm. Medical devices only cause harm if a sequence of events occur that results in a hazardous situation, which then causes or leads to harm.
For each identified hazard, consider the reasonably foreseeable sequences or combinations of independent events that can result in a hazardous situation, and document the resulting hazardous situation(s).
A sequence of events can be initiated in all phases of the product’s life cycle, e.g., during transport, storage, installation, maintenance, routine inspection, decommissioning, and disposal. It is important to consider all applicable phases.
Risk analysis includes the examination of different sequences or combination of events related to a single hazard that can lead to different hazardous situations.
In order to identify foreseeable sequences of events, it is helpful to consider the events and circumstances that can cause them, often referred to as initiating or trigger events. A failure mode identified in an FMEA can be a trigger event that leads to a hazardous situation.
See Figure 2 below for a pictorial example between the relationship of sequence of events, hazard, hazardous situation, and harm. It is good practice and helpful to also document the foreseeable sequence of events that led to the hazardous situation, along with the events that triggered them.
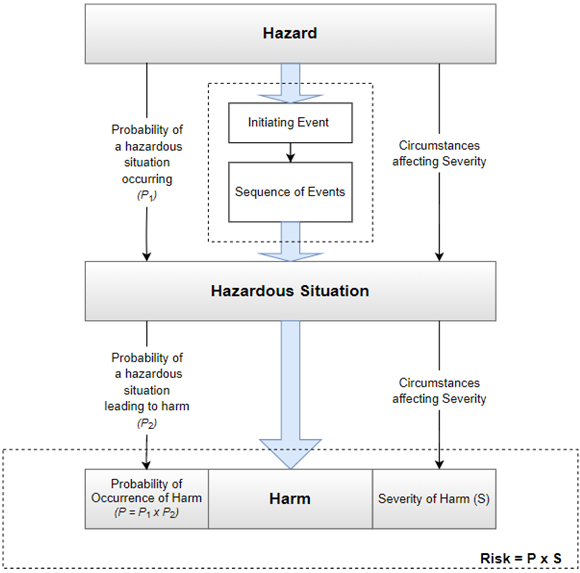
Figure 2: Relationship between hazard, hazardous situations, and harm
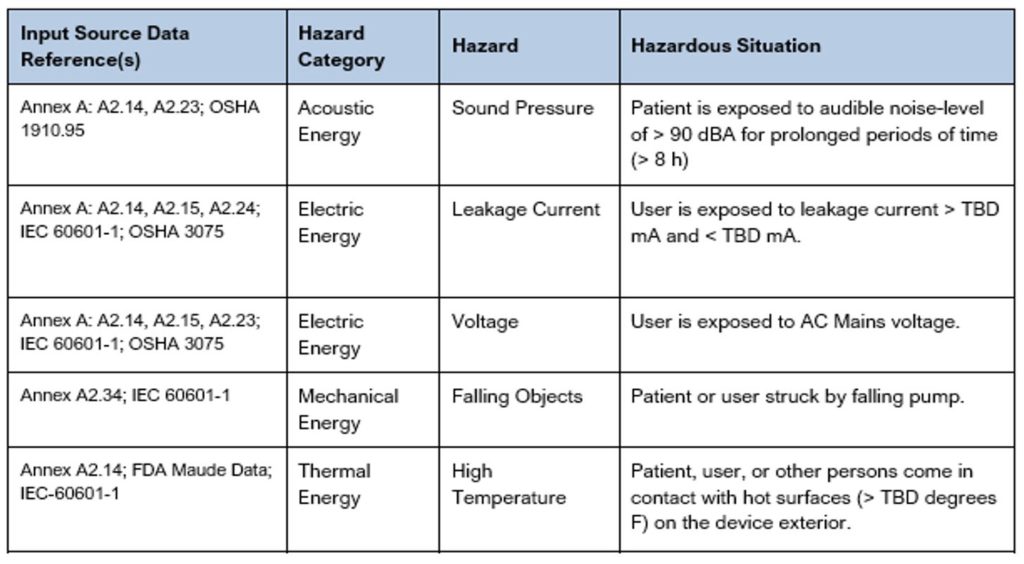
The hazardous situations identified/documented in a Preliminary Hazard List are often more generic until the details of the triggers and sequence of events are well defined. See Table 2 below that shows several examples of hazards and hazardous situations. As you can see in these examples, some of the hazardous situations have been left with TBDs until more is understood. For additional examples, reference the Sample Preliminary Hazards List created for an Infusion Pump. This sample document includes many more examples with different types of hazards and hazardous situations which we hope you find helpful as you step through the process.
Table 2: Excerpt from a Sample Preliminary Hazards List
NOTE: Each hazardous situation can lead to different types of harm.
- If multiple hazardous situations are possible for a given hazard, list each one on a separate row of the Product Risk Analysis.
- If multiple harms are possible for a given hazardous situation, list each one on a separate row of the Product Risk Analysis.
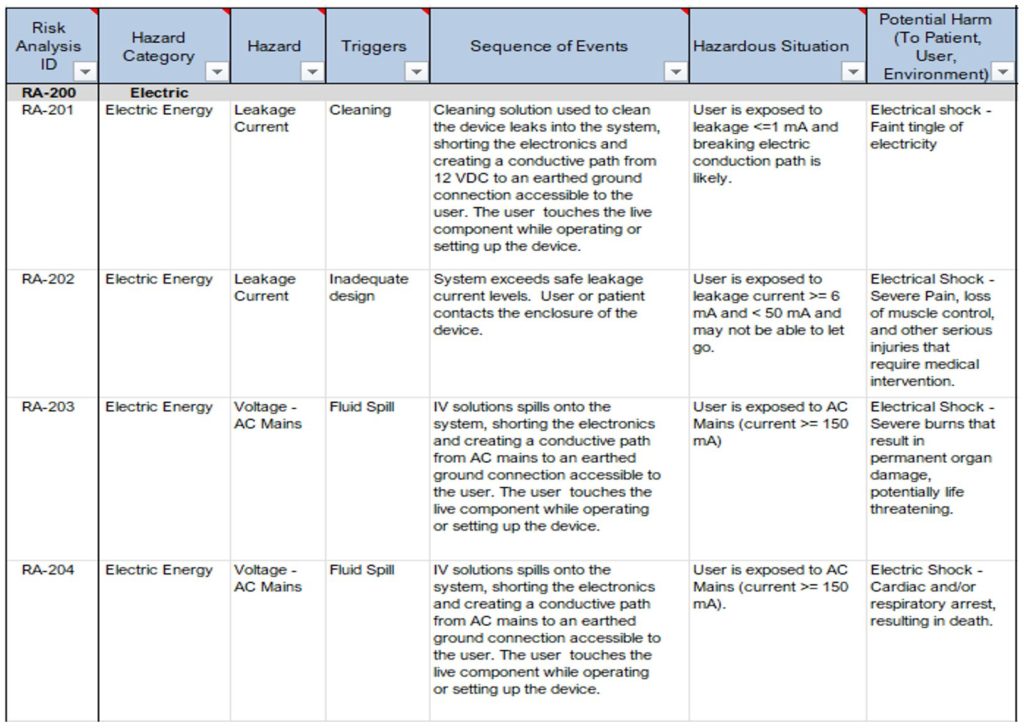
See Table 3 below for examples showing the relationships between hazards, triggers, foreseeable sequences of events, hazardous situations and the potential harm that can occur.
In the examples provided in Table 3, notice the following:
- Risk RA-201 and RA-202 have the same hazard “Leakage Current” but have different hazardous situations and different harms.
- Risk RA-203 and RA-204 have the same hazardous situation “User exposed to AC Mains (current>=150mA)” but have different harms associated.
For additional examples please see the Sample Product Risk Analysis created for an Infusion Pump. This sample document includes many more examples with different types of hazards, hazardous situations, harms, etc., and takes you through the whole process of documenting a Product Risk Analysis, which we hope you find helpful as you step through the process.
Table 3: Excerpt from a Sample Product Risk Analysis (Hazard -> Hazardous Situation -> Harm)
4. Risk Estimation
For each hazardous situation, estimate risk by determining the probability (P) of occurrence of harm and the severity (S) associated with that harm. In order to determine probability and severity, you should utilize all available information and data to inform your estimates.
The following are examples of information or data that can be used in estimating risk:
- Published standards
- Scientific or technical investigations
- Field data from similar medical devices already in use, including publicly available reports of incidents
- Usability tests employing typical users
- Clinical evidence
- Results of relevant investigations or simulations
- Expert opinion
- External quality assessment schemes for in vitro diagnostic medical devices
Probability of Occurrence
The probability (P) of occurrence of harm can be decomposed into two components (P1) and (P2) as described below:
- Probability (P1) that a hazardous situation occurs
- Probability (P2) that a hazardous situation leads to harm
It is not required to decompose (P) into its components, but it can be very helpful in estimating probability of occurrence of harm. You might find that you have a very high probability that a hazardous situation will occur, but the likelihood of it leading to harm is extremely rare, or vice versa. Breaking the probability of occurrence of harm into its components may help obtain better estimates.
Probability of occurrence can be quantitative or qualitative depending upon the situation. When sufficient data is available to estimate the probability of occurrence of harm with adequate confidence, the quantitative method is best to use. When such data does not exist, the qualitative method based on expert judgement is typically preferred.
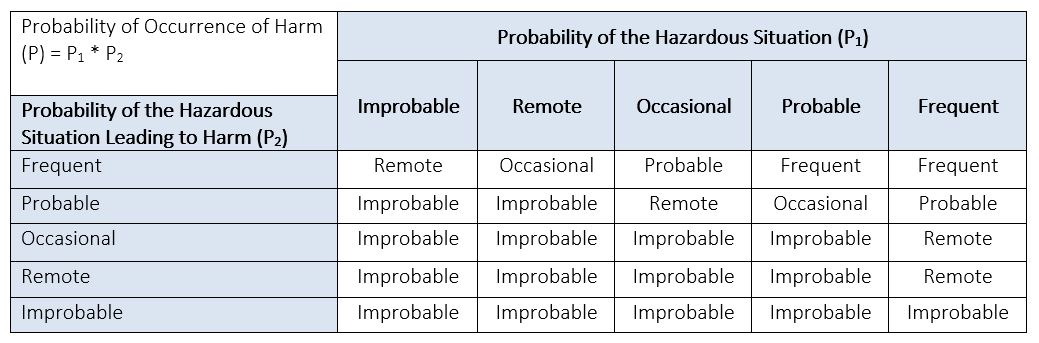
See Table 4 below for an example of a Probability of Occurrence Table. If you decide to break (P) into (P1) and (P2), you can modify this table to represent each component. A Probability of Harm Lookup Table, like the example provided in Table 5 below, can then be used to derive the probability (P) of occurrence of harm from its two components: (P1) and (P2).
These example tables, typically located in the Risk Management Plan, are used for risk estimation, and should be adjusted to be applicable to the particular type of product being analyzed.
Table 4: Example of Probability of Occurrence
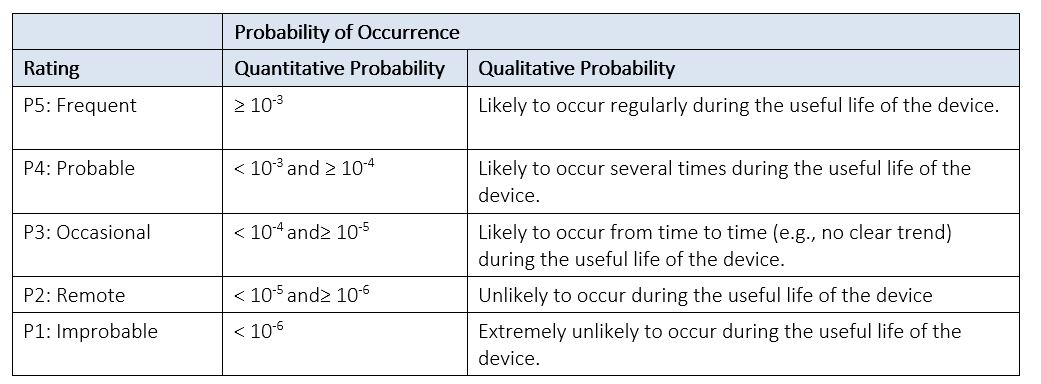
Table 5: Example of Probability of Harm Lookup Table
Severity
Severity (S) is the measure of the possible consequences of harm and is categorized using descriptors for the medical device for the purposes of risk estimation. See Table 6 below for an example of a severity table for a medical device.
Table 6: Example of Severity of Harm
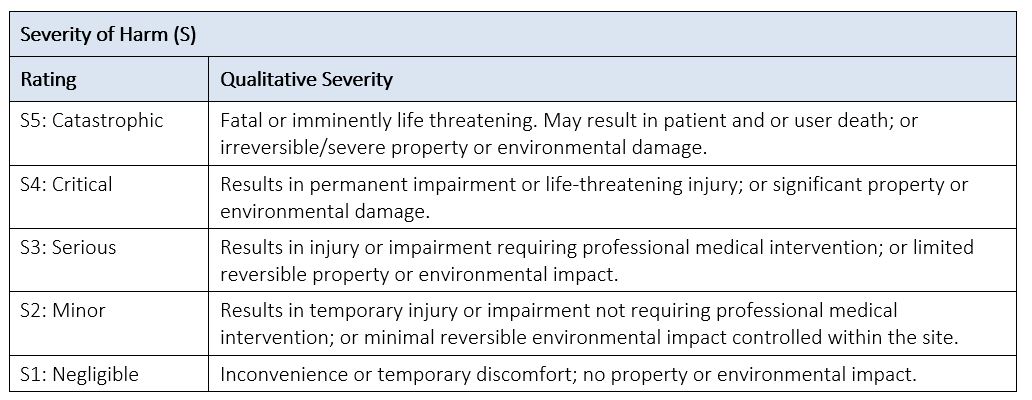
Severity levels should be defined with sufficient specificity so the correct level of severity can be assigned to the specific harms associated with the product.
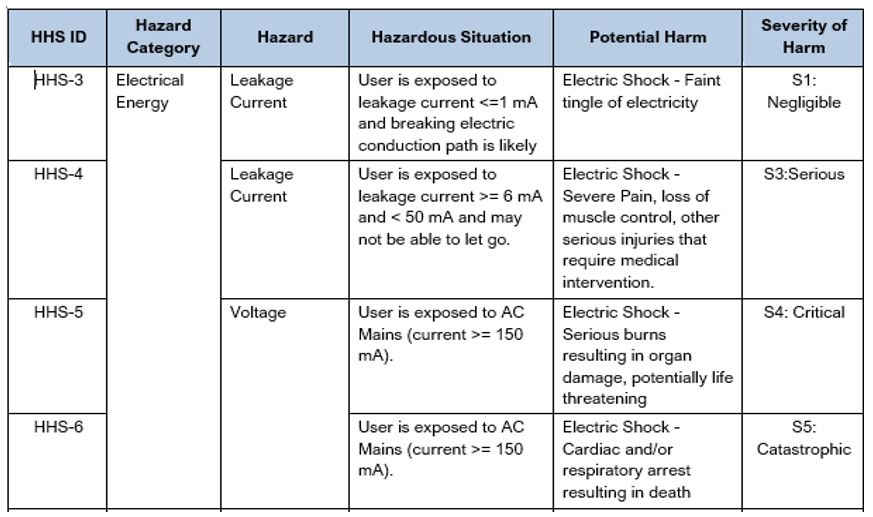
It is best practice to create a Hazard-Harm-Severity (HHS) table that is specific to your particular device. An HHS table associates the appropriate severity level for a particular hazardous situation. See Table 7 below showing several examples of linkages of Hazard-Harm-Severity. For additional examples see the Sample Hazard-Harm-Severity Table created for an Infusion Pump. Like the Sample Product Risk Analysis, this document has many more examples with different types of hazards, hazardous situations, harms, and severities which we hope you find helpful as you step through the process.
When conducting a Product Risk Analysis, referencing the HHS will help to ensure consistency, in the application of severity, based on agreed upon and approved clinical/application knowledge. You might choose to include this table in the Risk Management Plan for your device or release it as a separate document.
Table 7: Excerpt from a Sample Hazard-Harm-Severity (HHS) Table
Risk Level Matrix
It is helpful to define a Risk Level Matrix to allow for qualitative assessment of individual risks. A Risk Level Matrix is defined based upon the Probability of Occurrence of Harm levels and the Severity of Harm Levels. See Figure 4 below for an example of a typical 5×5 risk matrix with only 3 risk levels (Low, Medium, and High). A Risk Level Matrix is intended as a means of assessing the relative magnitude of risk associated with a hazardous situation and may be used along with the criteria for risk acceptability to determine if risk reduction is required. This matrix should be altered to suit the particular medical device.
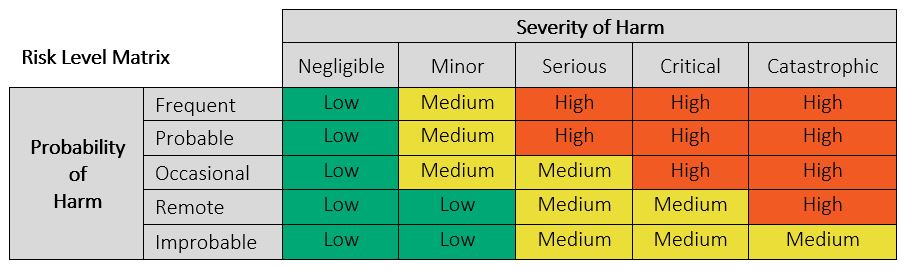
Figure 4: Example of 5 x 5 Risk Matrix
See Table 8 below for examples showing the next steps in estimating and evaluating the pre-mitigation risk for the hazards “Leakage Current” and “Voltage”. The risk was estimated using Table 4 for Probabilities, Table 7 for severity (from the HHS Table) and Figure 4: Example of 5 x 5 Risk Matrix.
Table 8: Excerpt from Sample Product Risk Analysis – Pre-Mitigation Risk Evaluation
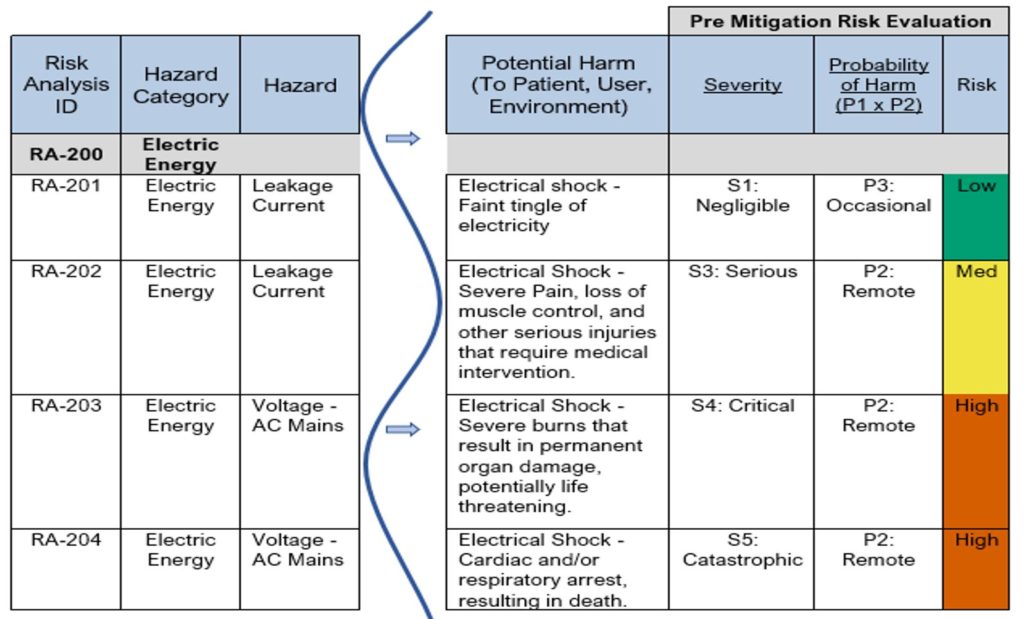
5. Evaluation of Risk
Evaluating risk means to determine whether risk is acceptable based upon pre-established criteria. Risk is to be evaluated for each identified hazardous situation.
- If the risk is deemed acceptable, further risk reduction may not be required depending on the approach to risk control identified.
- If the risk is not acceptable, risk control activities will be required to eliminate or reduce risk to an acceptable level.
The organization should define and document the criteria for risk acceptability based upon a risk control approach. ISO 14971 identifies several different acceptable approaches for risk control which include: reducing risk as low as reasonably practicable (ALARP), reducing risk as low as reasonably achievable (ALARA), reducing risk as far as possible (AFAP) without adversely affecting the benefit-risk ratio, and reducing risk based upon the magnitude of residual risk. Below are two examples demonstrating different criteria for risk acceptability using different approaches to risk control.
In Example 1 below, the criteria for risk acceptability are based on a risk control approach where risks are reduced depending on the magnitude of the residual risk.
Example 1: Risk Acceptability based on Magnitude of Residual Risk
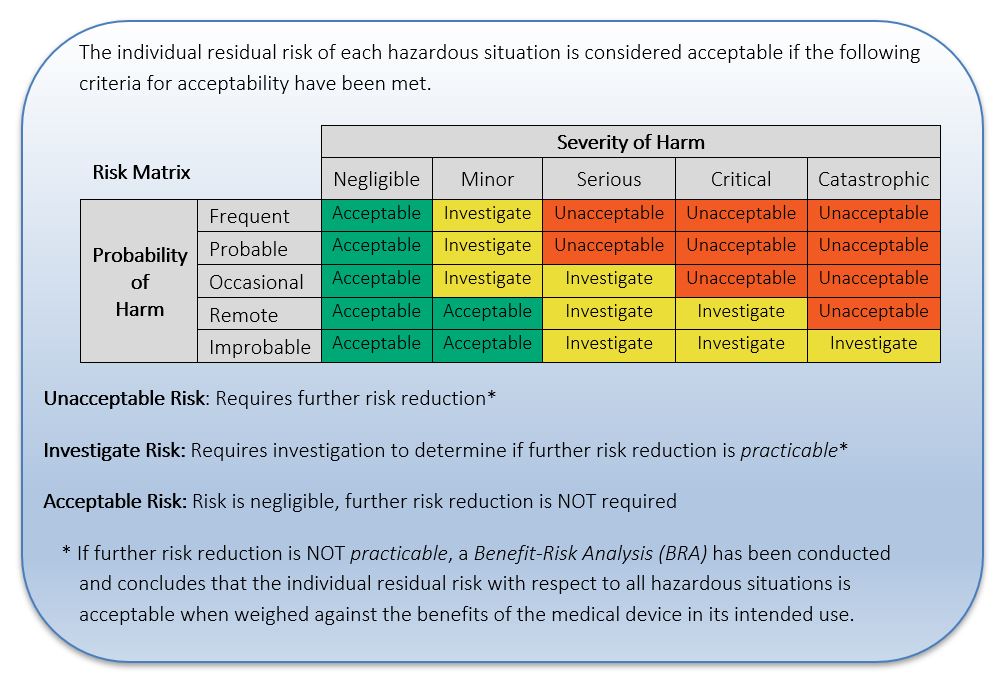
In Example 2 that follows, the criteria for risk acceptability are based on a risk control approach where risks are reduced as far as possible (AFAP). Consideration is given to whether technically practicable measures would reduce the risk without impacting the intended use or the benefit of the medical device.
NOTE: This approach to risk control is a requirement for compliance with EN ISO 14971:2019+AMD11:2021, which is particularly important if you intend to sell a product in Europe.
One can use a Risk Matrix, as with Example 1 above, but the relative magnitude of the risk has no direct impact on the criteria for risk acceptability; every risk, no matter the magnitude, is required to be reduced as far as possible.
Example 2: Risk Acceptability Criteria based on AFAP

6. Identify Risk Controls
When risk control measures are required, one or more of the three risk control options listed below should be used to reduce risk. The options below are listed in the order of their effectiveness, with Design being the most effective. It is not recommended to rely upon Labeling as your only option for risk control.
Design (D) – Inherently safe by design. Making the medical device design inherently safe by:
- Eliminating a particular hazard (e.g., eliminating sharp edges that can cause injuries),
- Reducing the probability of occurrence of the harm (e.g., reducing probability of fibrillation harm due to an electric shock by having no accessible live parts), or
- Reducing the severity of the harm.
Protective (P) – Measures taken in the medical device itself or in the manufacturing process. Taking protective measures by:
- Preventing the occurrence of a hazardous situation (e.g., automatic cut-off circuits, guards/covers, inspection/testing to detect non-conforming products), or
- Preventing a hazardous situation from leading to harm (e.g., visual or acoustic alarms to alert the user to a hazardous situation)
Labeling (L) – Information for safety and, where appropriate, training to users. Providing information for safety by:
- Placing warnings on the medical device,
- Including contraindications in the accompanying documentation,
- Providing instructions to support correct use and to avoid user error,
- Providing instructions to use personal protective equipment (e.g., use gloves and eyeglasses when handling toxic or hazardous chemicals and gases),
- Providing instructions about measures to reduce the severity of harm (e.g., rinse immediately with water after contamination with hazardous substances),
- Providing training to users on how to use the medical device correctly, and
- Providing instructions related to installation and maintenance during the lifetime of the medical device (e.g., maintenance intervals, maximum expected lifetime, how to clean, how to dispose, etc.)
Application of relevant standards, as part of the medical device design criteria, might constitute risk control activities. For hazards and hazardous situations that are fully covered by an international safety standard (e.g., IEC 60601-1, etc.), the manufacturer can often rely upon meeting the requirements of that standard to demonstrate acceptable risk.
In some cases, it may be determined that there is no practicable way of reducing risk to acceptable levels according to the criteria for risk acceptability documented. In these situations, a benefit-risk analysis should be carried out to determine whether the benefits of the medical device outweigh the residual risk. This step is important in order to show that every effort was first made to reduce risks to the pre-established acceptable levels. If the method of risk reduction chosen was AFAP, then a benefit-risk analysis is already a requirement for acceptability.
All risk control measures identified should be directly tied to a design requirement and/or specification. As new control measures are identified, review and update the appropriate requirement documents.
7. Implementation and Verification of Risk Control Measures
Risk control measures must be implemented and verified, where verification includes two distinct activities as summarized below:
Verification of Implementation
Verification of implementation of a risk control measure in the medical device, including information for safety, can be obtained by checking design documentation.
Verification of Effectiveness
Verification of effectiveness of risk control measures in the medical device, including information for safety, can be obtained by verification and validation of the medical device and accompanying documentation to ensure that the risk controls meet their associated design input requirements and prevent harm or damage.
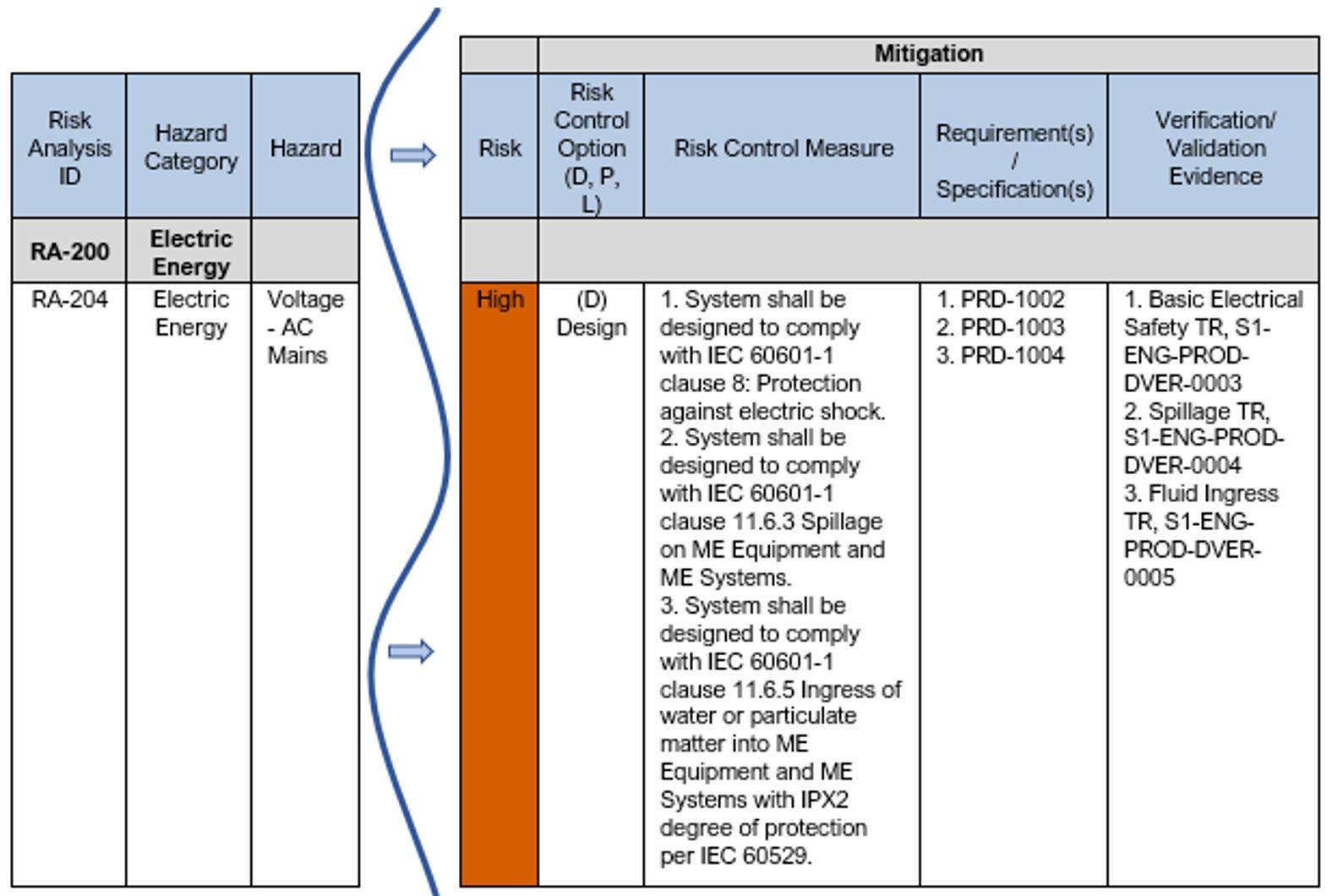
See Table 9 below for an example showing the next steps in documenting the mitigation to reduce the risk for a hazard. Note: in this example the criteria for risk acceptability being used is per Example 1: Risk Acceptability based on Magnitude of Residual Risk. Per this risk acceptability criteria, the High risk level = “Unacceptable” and risk reduction is required.
As you can see in this example, the risk control option selected is “Design”. The risk control measures are defined and trace to requirements and the requirements trace to the verification test evidence.
Table 9: Excerpt from Sample Product Risk Analysis – Mitigation
8. Residual Risk Evaluation
After control measures have been implemented and verified, evaluate the residual risk (i.e., the remaining risk) using the same method and criteria for risk acceptability used during the initial evaluation of risk in step 5 above.
If further risk reduction is NOT practicable, a Benefit-Risk Analysis (BRA) will be required to determine whether the benefits of the medical device outweigh the individual residual risk.
Note: If the approach to risk control is AFAP, then a Benefit-Risk Analysis (BRA) is already required for all risk in order to determine acceptability.
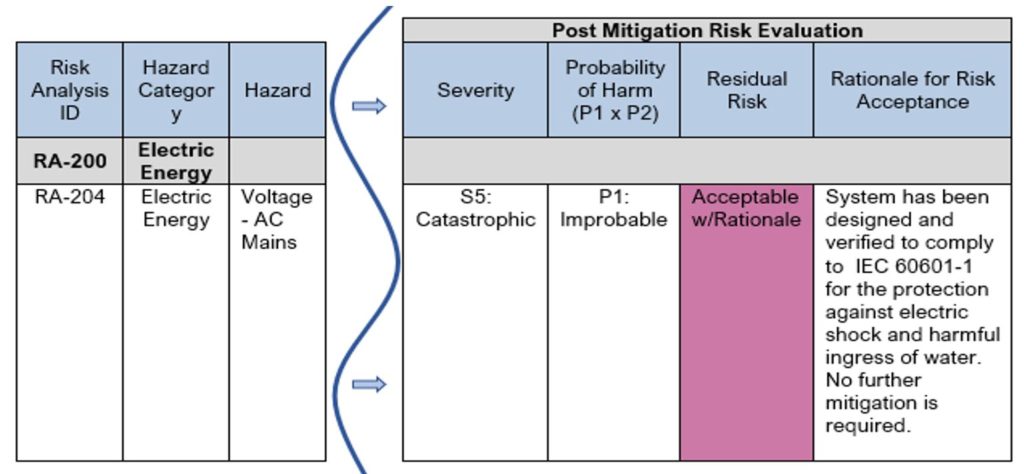
See Table 10 for an example showing risk evaluated after implementation and verification of the control measures. This example is a follow-on from Table 9 above, where the design mitigation for the “Voltage” hazard makes the medical device inherently safe by reducing probability of occurrence of harm due to an electric shock by designing to IEC 60601-1, a basic safety and essential performance standard for Medical electrical equipment.
In the risk estimation of this particular hazard post-mitigation, the severity remains the same (Catastrophic), but the Probability reduces (from Remote to Improbable).
Using the risk matrix in Figure 4, and the criteria for risk acceptability per Example 1: Risk Acceptability based on Magnitude of Residual Risk, we find the residual risk remains a “medium” risk, which would require further risk reduction or a Benefit-Risk Analysis. However, because this hazard and hazardous situation is fully covered by the international safety standard IEC 60601-1, we can rely upon meeting the requirements of that standard to demonstrate acceptable risk. As you can see in Table 10 below, the residual risk is identified as “Acceptable w/Rationale” and the rationale is provided for in the next column.
Table 10: Excerpt from Sample Product Risk Analysis – Post Mitigation Evaluation
9. Risks Arising from Risk Control Measures
Review each risk control measure to determine if it can be a source of risk. All risk control measures should be reviewed to determine whether:
- New hazards or hazardous situations are introduced, or
- The estimated risk for previously identified hazardous situations are affected by the introduction of risk control measures.
Any new or increased risks should be managed just like all the previous risks identified, starting with estimating the risk per step 4 in Table 1 above.
10. Benefit-Risk Analysis
When residual risks are NOT judged acceptable per the pre-established acceptance criteria and further risk reduction is not practicable, a benefit-risk analysis is performed to determine if the expected benefits of the intended use of the medical device outweigh the residual risk.
The decision as to whether benefits outweigh risks is a matter of judgement by experienced and knowledgeable individuals, usually comprising medical, clinical, or application experts.
If the evidence does not support the conclusion that the benefits outweigh the residual risk, the team may need to consider modifying the medical device or its intended use. Otherwise, the risk remains unacceptable, and the device should not be put on the market as designed.
11. Risk Completeness
Upon completion of the risk control activities, a review should be held to ensure that the risks from all identified hazardous situations have been considered and all risk control activities are completed. These reviews are a critical part of the process, and the results are to be included in the Risk Management File for the product.
CONCLUSION
Conducting a product risk analysis according to the requirements of ISO 14971 will not only help to ensure you develop a safe product and reduce your liabilities, but it will also help to ensure you can meet risk requirements for FDA submission of a medical device and other regulatory bodies, as well as satisfy risk requirements for CE Marking.
Risk analysis is most effective when it starts in the earliest phase of the product development process. The beauty of a risk analysis is that detailed design is not needed to begin identifying hazards and hazardous situations associated with a product, you just need to know what the product is and what it is intended to do. The earlier a risk analysis is started, the earlier high-risk areas of the design can be identified, allowing a change in architecture /design paths before the design is more rigid and more costly to change.
It is important to remember that a product risk analysis is not a singular activity; it will require updates throughout product development, production, and post-production as new information is provided regarding new hazards, hazardous situations, probabilities, and severities. Proposed changes to a medical device and/or its manufacturing processes should be evaluated for their effects on the safety of the use of the device. Some of these changes can introduce new hazards, eliminate existing hazards, or change probabilities or severity levels, thereby changing the level of risk associated with a hazard. If a change takes place, the current product risk analysis should be reviewed and updated as necessary.
We hope you will find these best practices useful in helping guide your next risk analysis and encourage you to leverage the companion templates as well as the sample documents listed in APPENDIX A to help you through the process.
APPENDIX A – TEMPLATES AND SAMPLE DOCUMENTS
Templates
- Preliminary Hazards Analysis Template
- Product Risk Analysis Template
Sample Documents
- Sample Preliminary Hazards Analysis
- Sample Hazard-Harm-Severity Table
- Sample Product Risk Analysis
*All templates and sample documents mentioned above are available for download on our Robust Product Risk Analysis Templates page. Simply enter your email address in the pop-up that appears on your screen and we will email them all to you!
APPENDIX B – COMPARISON BETWEEN RISK ANALYSIS AND FMEA
A risk analysis is required by the FDA for regulatory submissions, as well as by other countries in order to obtain necessary markings and registrations to release products to market. An FMEA, on the other hand, is NOT an explicit requirement of any regulatory body. The key differences between the two analyses are summarized in the table below.
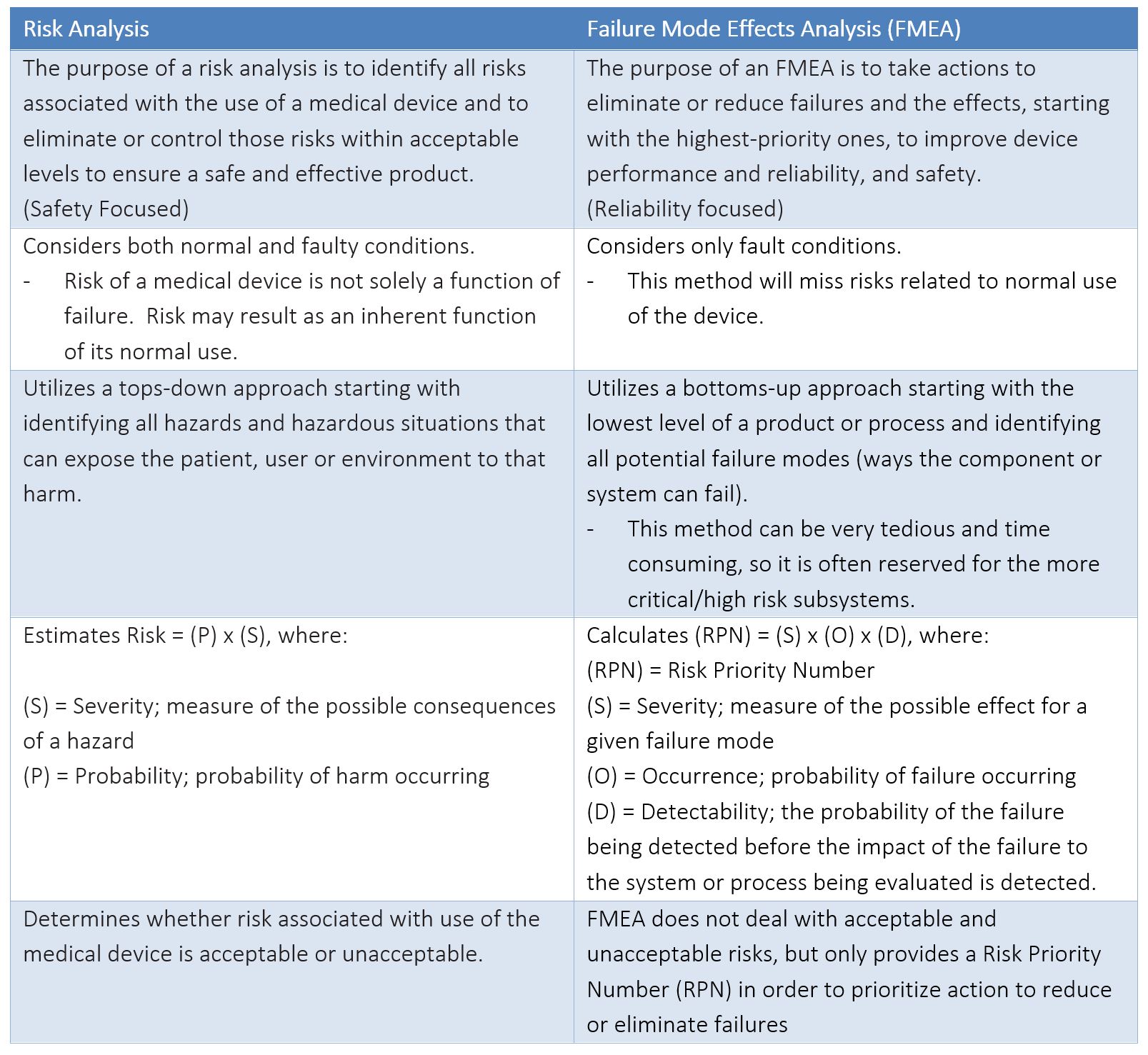
The FMEA is a great tool for identifying specific failure modes to improve reliability of a product. The FMEA is also a tool used to support a risk analysis; the failure modes identified by FMEA could act as a trigger event that could result in a hazardous situation exposing a patient, user, or environment to harm. Often failure modes identified in an FMEA will be linked to the Hazard identified in a Product Risk Analysis.
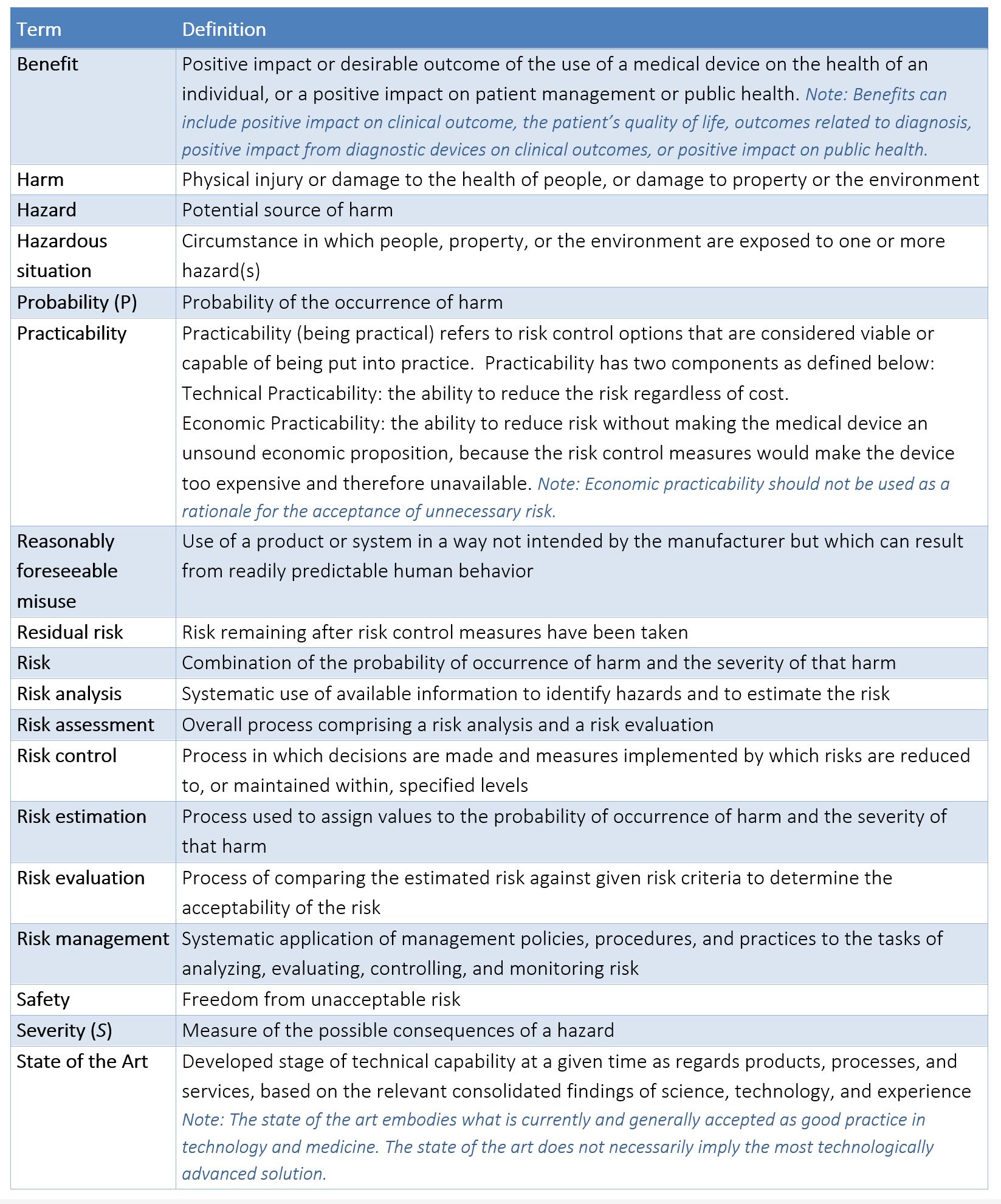
APPENDIX C – TERMS/DEFINITIONS AS DEFINED BY ISO 14971:2019
ABOUT THE AUTHOR
Theresa Ramirez is a Senior Quality Engineer at Simplexity Product Development working out of their San Diego office. Theresa has earned a BS degree in Bioengineering from UCSD. She is also an ASQ Certified Quality Engineer (2010) and Certified Medical Device Auditor (2021). Theresa has over 25 years of experience in product development, including 15+ years as a Quality Lead for medical device products.
